

大湾区大学物质科学学院及智能计算研究中心夏广杰研究员与浙江科技大学王亚敏博士合作,通过系统的密度泛函理论计算,深入揭示了轴向配体修饰对MN₄单原子催化剂电子结构与催化性能的调控机制,为理性设计高性能非贵金属氧还原催化剂提供了新的理论指导。该理论计算成果以“Axial Ligand Engineering: A Strategy for Promoting Oxygen Reduction Activity of MN₄ Single-Atom Catalysts”为题发表在催化领域知名期刊Applied Surface Science (IF 6.9)。
研究背景
氧还原反应是质子交换膜燃料电池等清洁能源技术的核心反应,其缓慢的动力学亟需高效催化剂。M-N-C单原子催化剂凭借高原子利用率和不俗的活性,成为替代贵金属铂的候选材料,但其性能仍面临挑战。近期一些实验偶然发现:在平面MN₄中心引入轴向配体是提升催化活性的有效策略,然而关于轴向配体如何影响电子结构、中间体吸附及反应动力学的微观机制仍有待深入探究。
研究结果
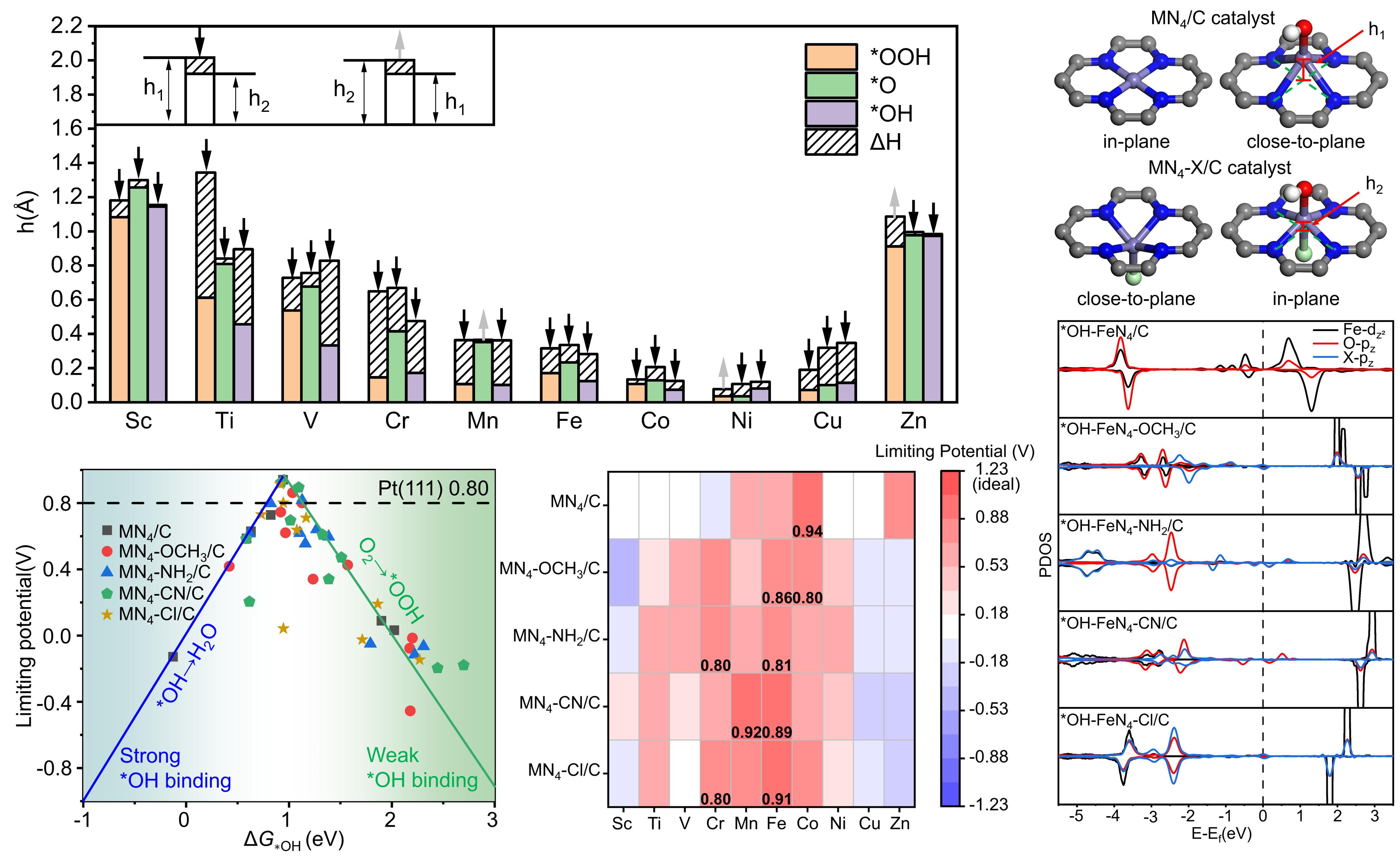
本研究通过密度泛函理论(DFT)计算和从头算分子动力学(AIMD)模拟,系统研究了超过50种轴向配体修饰的MN₄/X-C催化剂。结果表明,轴向配体的引入不仅能通过与反应中间体相反的拉力效应稳定金属中心,防止其在反应过程中过度脱离基底平面,更重要的是能直接调控金属中心dz²轨道的电子占据。对于活性提升最显著的FeN₄体系,我们通过理论计算发现,轴向配体与关键中间体OH竞争配位Fe的dz²轨道,这种电子竞争效应削弱了Fe与OH之间的过强相互作用,使*OH的吸附自由能更接近火山图顶峰的最佳值,从而显著提升反应活性。此外,在显式水溶剂环境下的分子动力学模拟进一步证实,轴向氯配体修饰的FeN₄-Cl/C催化剂在决速步*OH加氢步骤中具有更低的自由能垒,与实验观测到的高活性相符。
该研究从原子和电子层面深入阐释了轴向配体工程提升M-N-C单原子催化剂ORR性能的普适性机理,明确了配体与中间体对金属中心dz²轨道的“竞争配位”是性能优化的关键。这一认识将催化剂优化的研究视角从平面内调控延伸至轴向维度,展现了通过轴向配体进行电子结构精细调控作为一种潜在新策略的价值。该工作不仅对ORR催化剂设计具有重要指导意义,其所揭示的机制也有望推广至其他涉及氧中间体吸附/脱附的电催化反应中。
浙江科技大学王亚敏、罗博洋为本文共同第一作者,通讯作者夏广杰研究员为本工作提供了关键指导。
论文原文链接: https://doi.org/10.1016/j.apsusc.2025.164605













